Workflow Management
Reproducible science
- Whenever possible, automate all steps of analysis to keep a record of everything that is done and to avoid introduction of errors
- This makes it easy to:
- Avoid errors where different results are based on different versions of files
- Reduce effort required to correct errors or improve methods
- Adapt workflow to new datasets
- Promote data/knowledge sharing
- Crowdsource error correction/pipeline improvements
- This is all covered in detail here:
- Wilson, G., Bryan, J., Cranston, K., Kitzes, J., Nederbragt, L., & Teal, T. K. (2017). Good enough practices in scientific computing. PLoS Computational Biology, 13(6), e1005510
Bash:
- Hard coded file name:
bwa mem -t 8 GRCh38Decoy/Sequence/BWAIndex/genome.fa \
FastQ/sample1_R1.fastq.gz FastQ/sample1_R2.fastq.gz \
| samtools view -S -bo Mappings/sample1.bam -
samtools sort - o Mappings/sample1_sort.bam Mappings/sample1.bam
- Sample names as arguments:
for sample in $@
do
bwa_mem -t 8 GRCh38Decoy/Sequence/BWAIndex/genome.fa \
FastQ/${sample}_R1.fastq.gz FastQ/${sample}_R2.fastq.gz \
| samtools view -S -bo Mappings/${sample}.bam
samtools sort - o Mappings/${sample}_sort.bam Mappings/${sample}.bam
done
- Pass sample names to script for paralle execution:
cat SampleList.txt | xargs -n 1 qsub MappingPipeline.sh
- File Tests:
for sample in $@
do
if [ ! -f $BASEDIR/Mappings/${sample}.bam ]
then
bwa_mem -t 8 GRCh38Decoy/Sequence/BWAIndex/genome.fa \
FastQ/${sample}_R1.fastq.gz FastQ/${sample}_R2.fastq.gz \
| samtools view -S -bo Mappings/${sample}.bam
if [ ! -f Mappings/${sample}_sort.bam ]
then
samtools sort - o Mappings/${sample}_sort.bam Mappings/${sample}.bam
fi
done
for sample in $@
do
if test FastQ/${sample}_R1.fastq.gz -nt $BASEDIR/Mappings/${sample}.bam \
|| test FastQ/${sample}_R2.fastq.gz -nt $BASEDIR/Mappings/${sample}.bam
then
bwa_mem -t 8 GRCh38Decoy/Sequence/BWAIndex/genome.fa \
FastQ/${sample}_R1.fastq.gz FastQ/${sample}_R2.fastq.gz \
| samtools view -S -bo Mappings/${sample}.bam
fi
if test Mappings/${sample}.bam -nt Mappings/${sample}_sort.bam
then
samtools sort - o Mappings/${sample}_sort.bam Mappings/${sample}.bam
fi
done
- Check exit status of each command:
for sample in $@
do
if test FastQ/${sample}_R1.fastq.gz -nt $BASEDIR/Mappings/${sample}.bam \
|| test FastQ/${sample}_R2.fastq.gz -nt $BASEDIR/Mappings/${sample}.bam
then
bwa_mem -t 8 GRCh38Decoy/Sequence/BWAIndex/genome.fa \
FastQ/${sample}_R1.fastq.gz FastQ/${sample}_R2.fastq.gz \
| samtools view -S -bo Mappings/${sample}.bam
if [ $? -eq 0 ]
then
echo "Finished bwa_mem for $sample"
else
echo "bwa_mem failed for $sample"
exit 1
fi
fi
if test Mappings/{sample}.bam -nt Mappings/{sample}_sort.bam
then
samtools sort - o Mappings/{sample}_sort.bam Mappings/{sample}.bam
if [ $? -eq 0 ]
then
echo "Finished samtools sort for $sample"
else
echo "samtools sort failed for $sample"
exit 1
fi
fi
done
-
or use
set -eto exit script after a command fails (see here) -
What about:
- inconsistently named inputs (including fastq files in different folders)
- bash paramter substitution
find FastQ -name *.fastq.gz | sort | xargs -n 2 qsub MappingPipeline.sh
- requesting different resources for different steps of pipeline
- inconsistently named inputs (including fastq files in different folders)
Make
- Used to compile source code into binary
- Developed at a time when compilation was extremely resource-intensive
- Allows “nightly builds” where only modified code is re-compiled
- Builds a dependency tree from implicit wildcard rules
- Can be used to develop bioinformatics pipelines
- However:
- limited functionality and flexibility
- perl-level syntax opacity
- doesn’t support parallelization
Snakemake
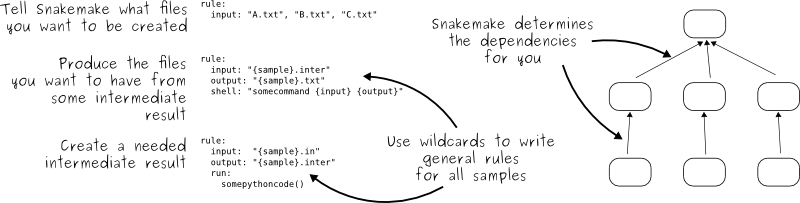
- Written in python
- Can be used to execute shell commands or python code blocks (in theory also R code blocks)
- Manages scheduling of job submission to cluster (or to the cloud)
- pe smp and h_vmem can be specified as params in the Snakefile or in a cluster config file
- cluster config files allow specification of default parameters
- Inputs, outputs, and parameters can be specified for each rule
- Need to include a “master rule” (usually called
all) which requires all of your desired outputs as input - Intermediate files can be automatically removed once they are no longer needed
- Supports benchmarking to report CPU and memory usage and Logging of messages/errors
- Supports config files to abstract details of pipeline from inputs and outputs
- Input functions allow config file entries to be accessed by wildcard values
- Workflows can also be further abstracted by:
- using
includestatements to import python code - using the
scriptcommand to execute a python script, giving it access to variables defined in the Snakefile - using
includestatements to import rules from other Snakefiles - creating sub-workflows
- using
- Conda environments can automatically be set up for each step of the analysis
- Many popular tools have prewritten wrappers that automatically create the necessary environment and run the tools using the specified inputs, outputs, and paramaters
- There is also a repository of example rules and workflows for NGS analyses
Getting started with Snakemake
- Snakemake documentation and tutorial
- Examples of:
- a Snakefile
- including additional Snakefiles
- a config file
- cluster configuration
- a bash script for invoking snakemake on the cluster, including email notification upon completion
Snakemake usage
- Do a dry run of workflow, printing commands to screen:
snakemake -np
- Produce a diagram of dependency tree:
snakemake -n --dag | dot -Tsvg > dag.svg
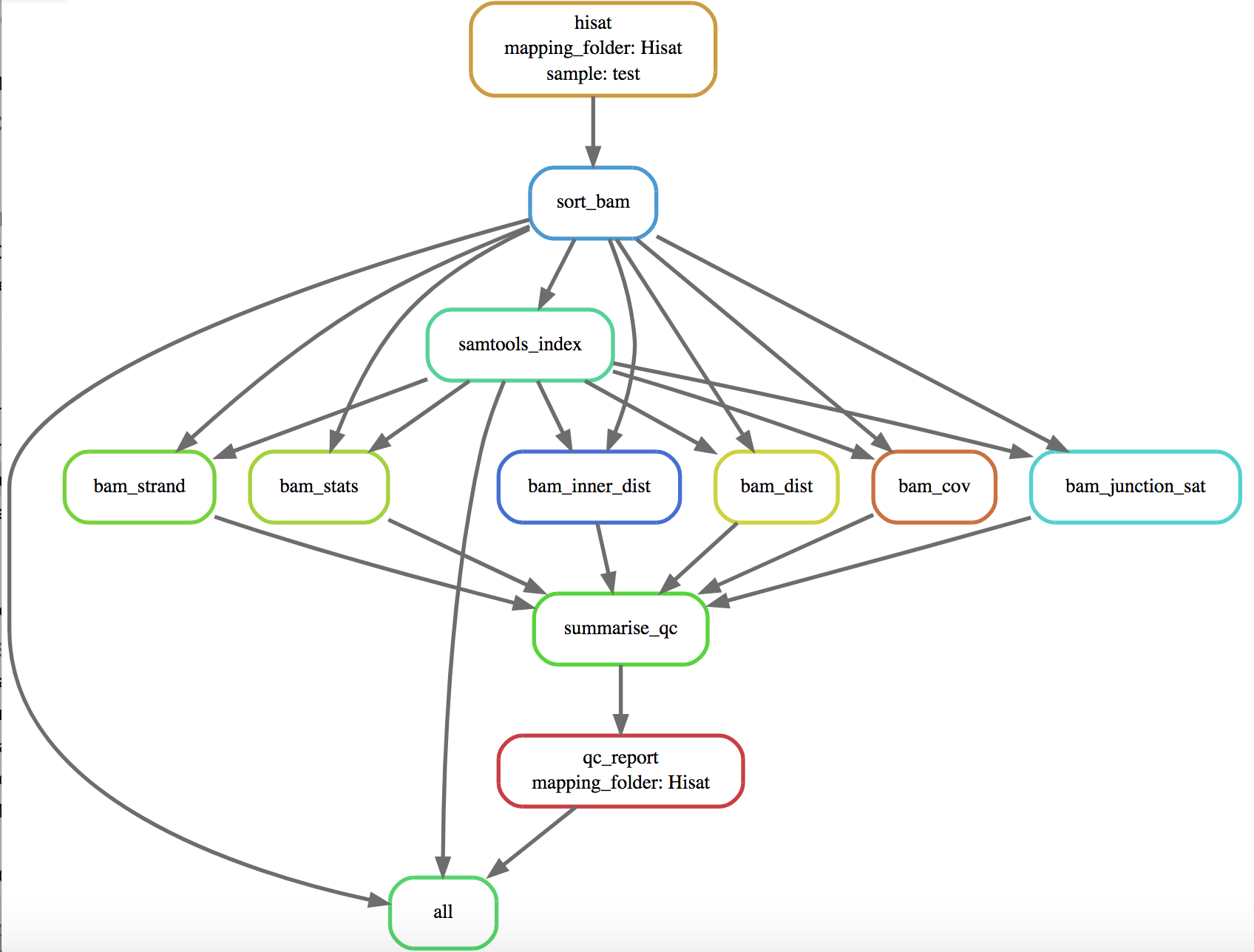
- Rerun rule (and all rules with it as a dependency):
snakemake -R RULENAME
- Rerun on new input files:
snakemake -n -R `snakemake --list-input-changes`
- Rerun edited rules:
$ snakemake -n -R `snakemake --list-params-changes`
- Submit jobs to cluster:
snakemake --use-conda --cluster-config cluster_config.yaml --cluster "qsub -pe smp {cluster.num_cores} -l h_vmem={cluster.maxvmem}" -j 20
- See here for additional command-line options
Alternatives to Snakemake
- Galaxy

- Common Workflow Language (CWL) / Docker
- See here for more

Workflow Management by Heath O’Brien is licensed under a Creative Commons Attribution 4.0 International License.